Activin A-Endothelin-1 Axis Drives Pulmonary Vascular Remodeling: Mechanistic Basis for ActRIIA Enzyme Activity Inhibitor Screening
1. Therapeutic Challenges in Pulmonary Hypertension and the Core Obstacle of Vascular Remodeling
Pulmonary hypertension (PH) is a critical condition characterized by progressive elevation of pulmonary artery pressure and extensive pulmonary vascular remodeling. Although current vasodilator therapies have achieved some progress, the long-term prognosis of patients remains fundamentally unimproved. Current treatment strategies primarily include endothelin receptor antagonists, prostacyclin analogs, and phosphodiesterase-5 inhibitors. While these drugs can alleviate clinical symptoms and improve hemodynamic parameters, they fail to fully halt or reverse structural vascular lesions. The persistent progression of vascular remodeling constitutes the core obstacle to long-term disease remission, indicating insufficient intervention in the underlying pathological processes by existing therapies.
Among the numerous vasoactive mediators involved in vascular remodeling, endothelin-1 (ET-1) is one of the most potent and persistently elevated key factors in PH. Elevated ET-1 levels are closely associated with disease severity and mortality, and its pathological effects extend far beyond vasoconstriction—ET-1 also drives oxidative stress, apoptosis, metabolic reprogramming, endothelial-mesenchymal transition, and impaired angiogenesis. Although endothelin receptor antagonists have become a cornerstone of PH treatment, their efficacy in improving underlying structural lesions remains limited, suggesting the existence of critical pathogenic pathways beyond ET-1.
2. Activin A: A Novel Driver of Vascular Remodeling in Pulmonary Hypertension
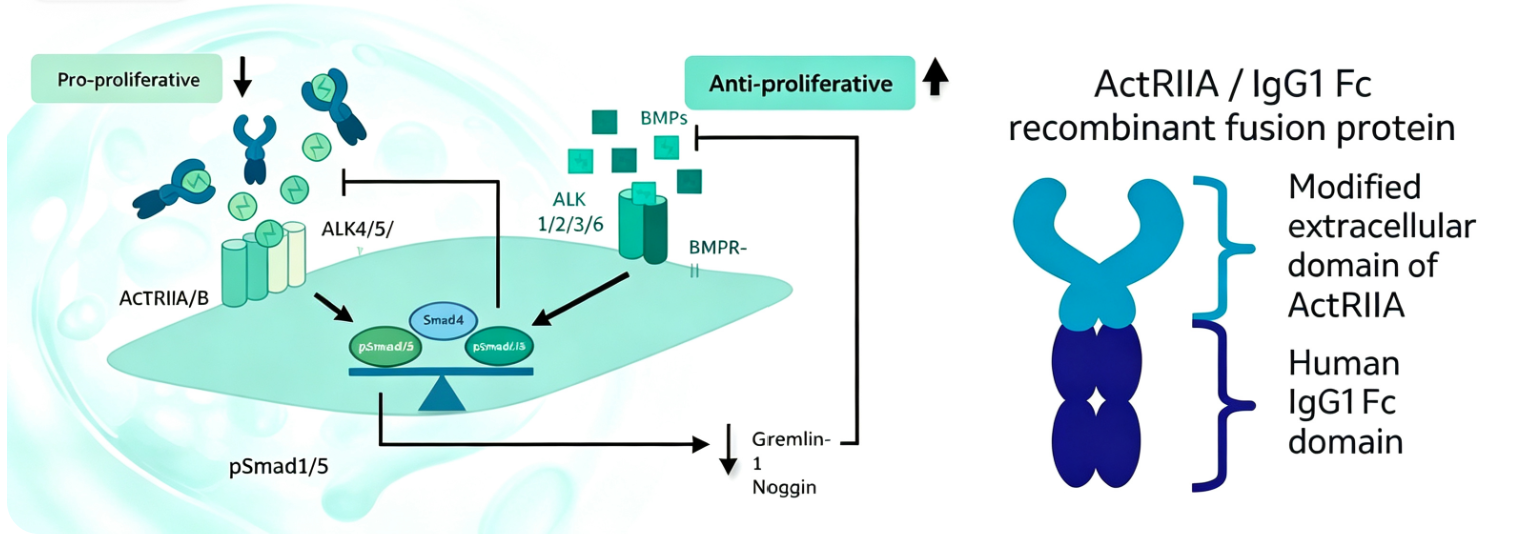
Recent studies have confirmed that endothelial-derived activin A overexpression is a key driver of vascular remodeling in PH. Activin A belongs to the transforming growth factor-β (TGF-β) superfamily and is composed of two inhibin βA subunits linked by disulfide bonds. It primarily exerts its biological effects by binding to type I and type II serine/threonine kinase receptor complexes. Research shows that activin A overexpression promotes endothelial dysfunction and accelerates the degradation of bone morphogenetic protein receptor type II (BMPRII), indirectly inhibiting the canonical BMPRII signaling pathway and exacerbating pulmonary vascular lesions.
Given that both activin A and ET-1 are endothelial-derived pro-remodeling mediators, researchers hypothesized that activin A may directly regulate the expression and activity of ET-1 in PH. A study published in *Arteriosclerosis, Thrombosis, and Vascular Biology* by the Kobe University Graduate School of Medicine systematically elucidated the molecular mechanisms of the activin A-ET-1 axis and its regulatory role in pulmonary vascular remodeling.
3. Activin A Upregulates ET-1 Expression via SMAD2/3 Signaling: Establishing the Upstream Regulatory Relationship
The study first validated the regulatory relationship between activin A and ET-1 in pulmonary artery endothelial cell models. Activin A stimulation or INHBA overexpression significantly increased ET-1 mRNA expression and protein secretion, whereas recombinant ET-1 treatment did not alter INHBA expression or activin A secretion, nor did the inflammatory factor TNF-α. These experiments established activin A as the upstream regulator of ET-1.
Mechanistically, activin A mediates ET-1 transcriptional upregulation through the canonical SMAD2/3 signaling pathway. The pharmacological inhibitor SB505124, which blocks SMAD2/3 phosphorylation, effectively suppressed activin A-induced ET-1 upregulation. Knockdown of SMAD2 or SMAD3 alone partially reduced ET-1 upregulation, while dual knockdown of SMAD2 and SMAD3 completely abolished the effect. This confirmed the synergistic and non-redundant roles of SMAD2 and SMAD3 in activin A-induced ET-1 expression.
4. Activin A-Driven ET-1 Mediates Multidimensional Pulmonary Vascular Pathological Changes
Activin A overexpression mediated multiple pulmonary vascular pathological changes through ET-1. In endothelial cells, activin A overexpression reduced eNOS expression and phosphorylation levels, impairing endothelial-dependent vasodilation. In a co-culture system of pulmonary artery endothelial cells and smooth muscle cells, activin A-stimulated endothelial cells significantly upregulated ET-1 and INHBA mRNA levels in smooth muscle cells, inducing a shift from a contractile to a synthetic/proliferative phenotype. This was characterized by increased PCNA and fibronectin expression and downregulation of contractile markers such as MYH11, SM22α, and α-SMA.
Additionally, activin A overexpression induced endothelial-mesenchymal transition, manifested by decreased VE-cadherin and increased fibronectin, vimentin, BMP4, SNAIL, and SLUG expression, along with increased co-localization of endothelial and mesenchymal markers. In terms of oxidative stress, activin A overexpression upregulated the central oxidative stress response protein NRF2, suppressed superoxide dismutase SOD2, and increased total reactive oxygen species production. Monocyte chemoattractant protein MCP-1 was also significantly elevated under activin A overexpression, suggesting that activin A may exacerbate vascular inflammation and remodeling by recruiting immune cell infiltration.
5. Dual-Pathway Inhibition Strategy: Therapeutic Potential Superior to Single ET-1 Blockade
The study further compared the functional effects of follistatin-mediated activin A inhibition versus bosentan-mediated ET-1 blockade. In INHBA-overexpressing endothelial cells, bosentan partially restored angiogenic capacity but failed to significantly reduce apoptosis. In contrast, follistatin (FST) alone or in combination with bosentan significantly improved tube formation and cell survival.
In vivo experiments using endothelial-specific INHBA-overexpressing mice and wild-type mice with chronic hypoxia-induced PH, FST-based treatment regimens were more effective than bosentan monotherapy in reducing right ventricular systolic pressure, improving right ventricular hypertrophy, and attenuating pulmonary vascular remodeling and medial thickening. Importantly, FST treatment significantly suppressed lung tissue ET-1 levels and corrected abnormalities in ET-1 downstream signaling. Notably, elevated IL-6 levels and increased macrophage infiltration in lung tissues during activin A overexpression were improved only by FST-based treatment, whereas bosentan monotherapy had no effect.
These findings collectively demonstrate that activin A is the master regulator of ET-1 production and its pathological effects in PH. Inhibiting activin A not only blocks ET-1-driven remodeling but also attenuates ET-1-independent pathogenic pathways, supporting its clinical potential as a more upstream therapeutic target.
6. ActRIIA Enzyme Activity Inhibitor Screening: Tools and Methods for Targeting Activin Signaling
The biological effects of activin A require binding to type II receptors (primarily activin receptor IIA, or ActRIIA) to initiate downstream signaling cascades. Thus, ActRIIA has become a key target for screening inhibitors of the activin signaling pathway. Inhibitor screening kits for this target typically employ chemiluminescent ELISA principles to evaluate the binding capacity of activin A to biotinylated ActRIIA and assess the inhibitory activity of candidate compounds. Such tools enable high-throughput screening, accelerating the discovery of drugs targeting the activin signaling pathway.
7. Which Suppliers Offer ActRIIA Enzyme Activity Inhibitor Screening Kits and Related Protein Products?
Nanjing U-Ability Biotech provides high-quality recombinant protein products, focusing on drug development, basic research, and cell therapy. They offer ActRIIA His Tag Protein, Human (Catalog No. UA080521), which can be used for activin signaling pathway research, ActRIIA inhibitor screening, protein interaction analysis, and drug target validation. This product is produced using the HEK293 eukaryotic expression system, ensuring high purity and strict endotoxin control standards, making it suitable for various biological functional studies.